Qu'est-ce que le syndrome de Marfan ?
Le syndrome de Marfan décrit un trouble héréditaire complexe du tissu conjonctif, affectant principalement les yeux, le système cardiovasculaire et le système musculo-squelettique. Cependant, étant donné que chaque organe est constitué de tissu conjonctif, le syndrome de Marfan peut idéalement détruire et fortement interférer avec la croissance et la fonction de tout site anatomique.
Le syndrome se transmet sur un mode autosomique dominant : nous sommes donc confrontés à une maladie génétique grave, ayant une « expression phénotypique » extrêmement variable (les défauts peuvent énormément différer d'une famille à l'autre ou d'un patient à l'autre).
Ce qui déclenche le syndrome de Marfan, c'est l'altération du gène FBN1 (sur le chromosome 15), qui code pour la fibrilline-1, une glycoprotéine conjonctive très importante qui constitue le support structurel des microfibrilles.
Microfibrilles : constituées de fibrilline, les microfibrilles sont présentes dans la matrice extracellulaire, dans laquelle elles forment un entrelacement pour le dépôt d'élastine dans les fibres élastiques.Bien qu'omniprésentes dans l'organisme, les microfibrilles abondent surtout dans l'aorte, les ligaments et les zonules du corps ciliaires (au niveau oculaire).
Comme il s'agit d'une maladie autosomique dominante, seuls les enfants qui ont hérité d'un gène FBN-1 altéré des deux parents sont touchés par le syndrome de Marfan. Néanmoins, dans un cas sur quatre, la maladie est le résultat de mutations spontanées chez des patients qui n'ont pas d'antécédents familiaux.
Le nom de la maladie vient du pédiatre français qui l'a décrite pour la première fois en 1896 (A. Marfan), après quoi il a fallu attendre 1991 pour identifier le gène altéré impliqué dans la manifestation symptomatologique : le découvreur était F. Ramirez.
Voir la vidéo
- Regardez la vidéo sur youtube
Causes
Nous avons mentionné que le syndrome de Marfan est l'expression immédiate de la mutation d'un gène qui code pour la fibrilline-1.
La fibrilline 1 est un composant glycoprotéique de l'élastine, essentiel pour assurer et maintenir l'élasticité et la résistance des tissus.Dans des conditions physiologiques, la fibrilline 1 se lie à une autre protéine, connue sous le nom de TGF-bêta (ou facteur de croissance transformant bêta). Le TGF-bêta semble être impliqué dans des processus délétères affectant le muscle lisse vasculaire et la matrice extracellulaire. Partant de ces hypothèses, certains auteurs sont convaincus que le syndrome de Marfan est dû, outre la mutation du gène FBN-1, également à un excès de TGF-bêta, notamment dans l'aorte, les valves cardiaques et les poumons.

Incidence
On estime que le syndrome de Marfan affecte 1 naissance sur 3 000 à 5 000 et survient sans distinction entre les hommes et les femmes. Les statistiques montrent que 75 % des patients ont des antécédents familiaux positifs ; dans les 25 % restants, la cause réside dans des mutations sporadiques qui semblent liées, d'une certaine manière, à l'âge avancé du père au moment de la conception.
Les enfants atteints de formes extrêmement sévères du syndrome de Marfan ont une « espérance de vie inférieure à un an ».
Avant l'évolution des stratégies chirurgicales à cœur ouvert, la plupart des patients atteints du syndrome de Marfan avaient une espérance de vie moyenne de 32 ans ; grâce à l'amélioration constante des thérapies médicales et pharmacologiques, les personnes actuellement atteintes du syndrome de Marfan vivent en moyenne jusqu'à 60 ans.
Signes et symptômes
Pour plus d'informations : Symptômes du syndrome de Marfan
Le syndrome de Marfan peut survenir de manière totalement asymptomatique. Les patients atteints ont une structure exagérément mince, étant disproportionnellement grands et minces. Les membres inférieurs et supérieurs sont beaucoup plus longs que le tronc (dolicosténomégalie). On parle aussi de arachnodactylie pour exprimer au mieux le concept de la longueur exagérée des doigts, typique des personnes atteintes du syndrome de Marfan : les mains sont donc comparées aux pattes d'une araignée.
En termes de taille, ces patients ont une stature avec une moyenne supérieure au 97e percentile.
Parmi les autres particularités souvent présentes chez les patients atteints du syndrome de Marfan, on retiendra également :
- Ouverture des bras supérieure à la hauteur
- Articulations lâches → mobilité articulaire exagérée
- Déformation de la paroi thoracique
- Déplacement de la lentille
- Haut du corps moins développé que le bas
- Pneumothorax spontané (11 %)
- Scoliose
- Stries cutanées au niveau de la cuisse, du dos, deltoïde, pectoral
Parmi les signes les plus problématiques associés au syndrome de Marfan, on retiendra le prolapsus de la valve cardiaque et l'insuffisance de la valve mitrale : une affection similaire peut facilement favoriser la dilatation de l'anneau aortique et la dissection aortique.
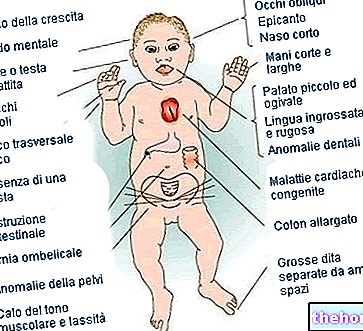
Le tableau montre les signes que l'on peut trouver chez les patients atteints du syndrome de Marfan. Les personnages qui y sont décrits ne sont pas toujours présents, mais on en retrouve une bonne partie.
Symptômes possibles
Peau
Stries dans la région thoracique, lombaire et sacrée
Yeux
Altération de la vision, astigmatisme, décollement de rétine, glaucome à angle fermé, luxation du cristallin, myopie
Structure osseuse
Arthralgie, cyphoscoliose, dolichosténomélie (développement excessif de la longueur des membres par rapport au tronc), hypermobilité, palais haut, poitrine déformée, pieds plats, poignets serrés et fins, rentrée/protusion anormale du sternum, scoliose, épaules courbes, spondylolisthésis
Les doigts
arachnodactylie
Poumons
Pneumothorax spontané, dyspnée, maladie pulmonaire obstructive idiopathique
Altérations du visage
Palais ogival (malformation du palais), rétrognathie mandibulaire (défaut de développement de la mâchoire), visage allongé
Cœur
Angine de poitrine, anévrisme de l'aorte abdominale, arythmie cardiaque, dilatation/rupture/dissection de l'aorte thoracique, insuffisance aortique, prolapsus de la valve mitrale
Langue
Difficulté avec la parole
Diagnostic
Compte tenu des plus de 200 mutations possibles, l'utilisation de marqueurs génétiques est presque impossible à des fins de diagnostic.
L'évaluation du syndrome de Marfan n'est pas toujours aussi immédiate, car l'expression phénotypique de la mutation n'est pas toujours évidente et facile à identifier. Le retard diagnostique peut compromettre gravement la survie du patient : il suffit de penser, par exemple, à la méconnaissance d'un problème cardiovasculaire.
Les critères diagnostiques du syndrome de Marfan ont été élaborés au niveau international en 1996 : le diagnostic consiste en « l'investigation de l'histoire familiale associée à une association de indicateurs majeurs et mineurs du syndrome.
Certains des nombreux tests de diagnostic utilisés sont :
- échocardiogramme
- angiorésonance magnétique et scanner (pour l'exploration de l'aorte)
- angiographie par résonance magnétique (ARM) avec produit de contraste (pour mettre en évidence les structures internes de l'aorte)
- examen aux lampes à fente (pour analyser l'éventuelle luxation du cristallin)
- mesure de la pression oculaire (pour mettre en évidence la présence éventuelle de glaucome)
- tests génétiques (recommandés avant de concevoir un enfant pour vérifier si oui ou non le syndrome)
Thérapies
Puisqu'il s'agit d'une maladie génétique, il n'existe aucun médicament ou traitement qui puisse inverser la maladie.
L'utilisation de médicaments est cependant indispensable pour soulager les symptômes et éviter toutes complications, notamment cardiaques.A cet effet, les médicaments pour diminuer la pression artérielle, comme les sartans (surtout), les IEC et les bêtabloquants, sont particulièrement adaptés.
Dans le cadre du syndrome de Marfan, les patients souffrant également de scoliose peuvent suivre un traitement spécifique, ainsi que pour ceux atteints de glaucome.
La chirurgie est envisageable pour corriger la dilatation aortique anormale, élément qui fédère souvent la majorité des patients atteints du syndrome de Marfan.
Continuer : Syndrome de Marfan - Médicaments et traitement "










-in-alto-abs.jpg)